


Background:
Myelodysplastic syndromes (MDS) have been associated with alterations in the bone marrow microenvironment and include abnormal innate and adaptive immune responses. The extent to which these changes contribute to the pathogenesis of MDS is unclear. The heterogeneous nature of MDS makes it difficult to distinguish changes in intrinsic cell function for microenvironmental alterations in the mutated myeloid compartment. T cells, which are in the lymphoid compartment and less frequently contain MDS mutations, can potentially be used to evaluate immune dysfunction in the presence of malignant myeloid cells. Here we examine the immunophenotypes and proliferative function of T lymphocyte and monocyte populations from higher-risk MDS patients.
Methods:
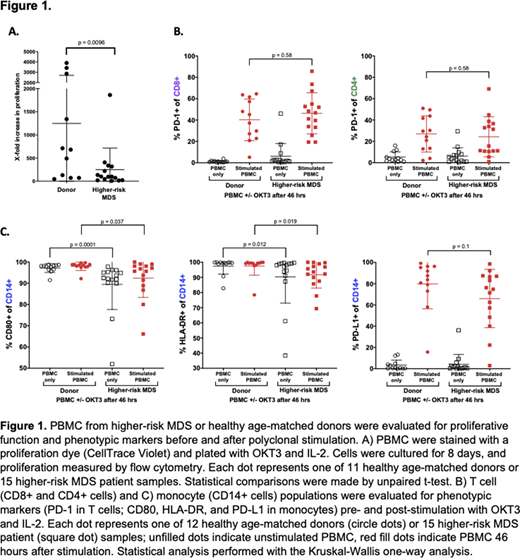
Peripheral blood mononuclear cells (PBMC) from patients with IPSS-R Intermediate/High/Very High-risk MDS (herein referred to collectively as higher-risk MDS) and age-matched healthy donors (n = 15 and n = 12, respectively) were polyclonally stimulated with an antibody against CD3 (OKT3) in the presence of IL-2 (50 U/mL). Phenotypic analysis was performed at baseline and 46 hours after the addition of OKT3 and IL-2. T cell proliferation was measured by staining PBMC with proliferation dye (CellTrace Violet) and measuring fold proliferation after eight days in culture. Proliferation statistical comparisons were made by unpaired t-test. Phenotypic parameters were measured by flow cytometry and statistical analysis performed with the Kruskal-Wallis one-way analysis.
Results:
As previously reported, higher-risk MDS patient T cells had decreased capacity for proliferation when compared to healthy donor counterparts (Figure 1A p = 0.0096). This difference in proliferative capacity was not associated with either the level of PD-1 on the surface of T cells (r = 0.2956) or the level of PD-L1 on monocytes (r = 0.3859) in stimulated PBMCs. Patients with higher-risk MDS tended to have lower PD-L1 surface expression on monocytes compared to healthy donor counterparts, albeit not significantly (Figure 1C p = 0.1). However, PD-1 expression on T cells was comparable between groups both before and after stimulation (Figure 1B CD4 cells: p = 0.58; CD8 cells p = 0.58). This suggests that T cell proliferation from patients with higher-risk MDS might not be inhibited through the PD-1/PD-L1 axis. Of note, higher-risk MDS patient monocytes expressed less CD80 and HLA-DR both pre- and post-stimulation when compared to healthy donors (Figure 1C CD80: p = 0.0001 and 0.037; DR: p = 0.012 and 0.019, respectively), implying a potential dysfunction in the monocytic stimulatory and antigen presentation capacity. PD-L1 surface expression is low at baseline (an average of <5% for both donors and higher-risk MDS); upon stimulation, there is a high correlation between HLA-DR and PD-L1 in healthy donors (p = 0.0003, r = 0.859). Additionally, there is less upregulation of PD-L1 on the surface of monocytic cells in the higher-risk MDS group and subsequently less correlation between PD-L1/HLA-DR (p = 0.03) in stimulated PBMC samples. Taken together, these results show a decreased baseline expression and/or ability to upregulate monocyte activation markers after stimulation. We hypothesize that these changes might contribute to the observation that higher-risk MDS patient T cells are less capable of proliferation.
Conclusion:
There was no significant difference in PD-1 and PD-L1 expression between higher-risk MDS and healthy donor patients, despite reduced T cell proliferation among patients with MDS. Additionally, decreased levels of CD80 and HLA-DR on intermediate/high-risk MDS patient monocytes suggests a suppressed ability to upregulate activation markers, possibly leading to decreased T cell proliferation. Targeting the PD-1/PD-L1 axis alone may not be therapeutic in patients with higher-risk MDS, and we hypothesize that the immune evasion observed in higher-risk MDS patients may not be due exclusively to increases in inhibitory interactions like PD-L1 and PD-1, but may result from a decrease in stimulatory receptors, such as CD80, and antigen presentation molecules, such as HLA-DR.
Fields:PersImmune, Inc.: Ended employment in the past 24 months. Ferrari:PersImmune, Inc.: Ended employment in the past 24 months. Tarke:PersImmune, Inc.: Ended employment in the past 24 months. Luger:Arcturus Therapeutics: Current Employment. Lane:PersImmune, Inc.: Current Employment. Vitiello:PersImmune, Inc.: Current equity holder in private company, Ended employment in the past 24 months. Bejar:Aptose Biosciences, Inc: Current Employment, Current equity holder in publicly-traded company.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal